What is gene phylogeny?
Constructing a Phylogenetic TreeBlast and Homologene within GenBank can provide sequences of protein homologs that can be downloaded and analyzed in Mega. Files with the FASTA file extension from these web tools in Mega can construct trees in a few different ways as outlined in the results section.
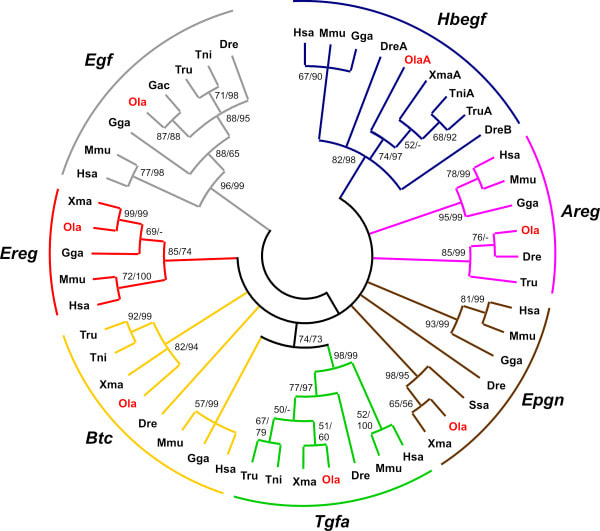
|
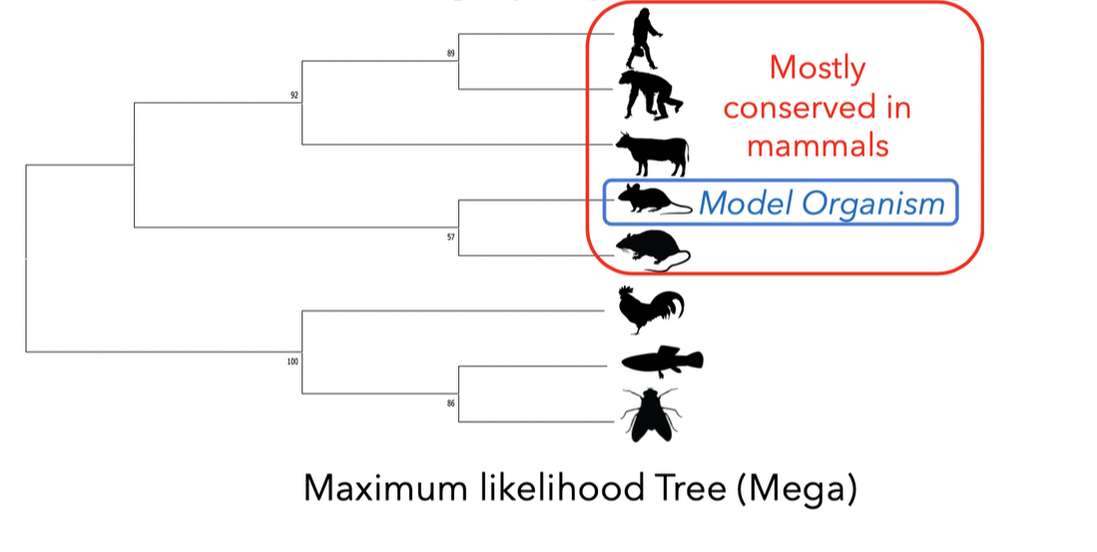
EGFR Ligand phylogeny is diverse, which makes signaling complex and interesting. To the left is a tree showing growth signaling receptor ligands similar to EGF. Their evolutionary relationship may be important to genetics-associated disease, of which LADC is. The numbers correspond to the likeliness that each element of the tree is evolutionarily related. Terminology such as "clade" can be used to describe a set of organisms all with one common ancestor [1]. For EGFR homology, mammalian species seem to exist in a separate well-conserved clade.
|
Results
Maximum Likelihood (ML)- ML evaluates all possible probabilities of predicted trees simultaneously. This statistical model determines likelihood given a set of observations, in this case amino acid sequences. This method is computationally complex, but often yields the most information. For the set of amino acids from common EGFR isoforms amongst the following organisms was generated using Neighbor Joining (NJ). The NJ method is generally rapid and accurate, but only produces a single tree and doesn't consider other options [2]. However, this method is especially useful for sets of organisms with highly varying lineages.
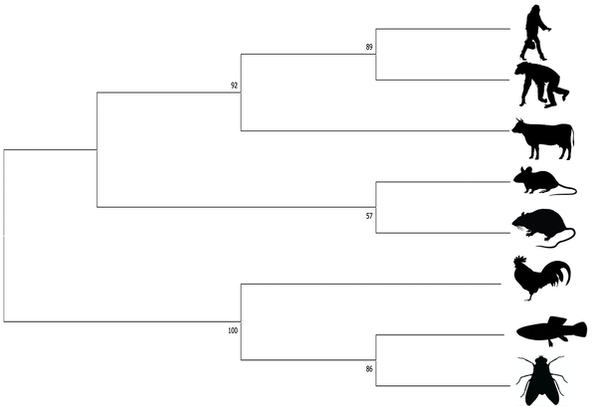
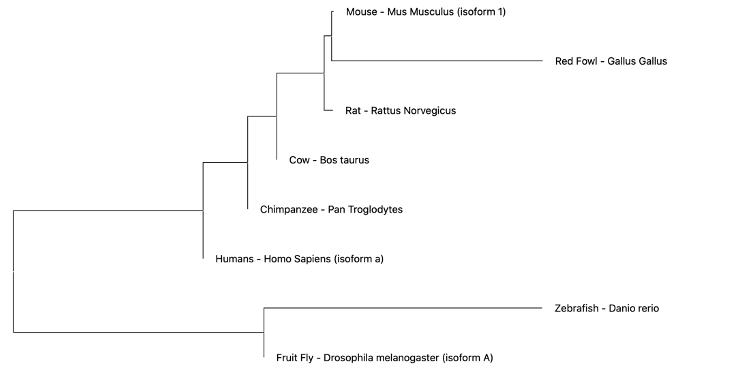
Neighbor Joining (NJ)- The NJ method is generally rapid and accurate, but only produces a single tree and doesn't consider other options. However, this method is especially useful for sets of organisms with highly varying lineages [2].
Minimum Evolution (ME)- The ME model minimizes the sum of branch lengths for predicted trees. It then optimizes the internal branch lengths to generate a final tree [3]. It generates minimal spanning trees, which are the shortest paths between boundary nodes.
Minimum Evolution (ME)- The ME model minimizes the sum of branch lengths for predicted trees. It then optimizes the internal branch lengths to generate a final tree [3]. It generates minimal spanning trees, which are the shortest paths between boundary nodes.
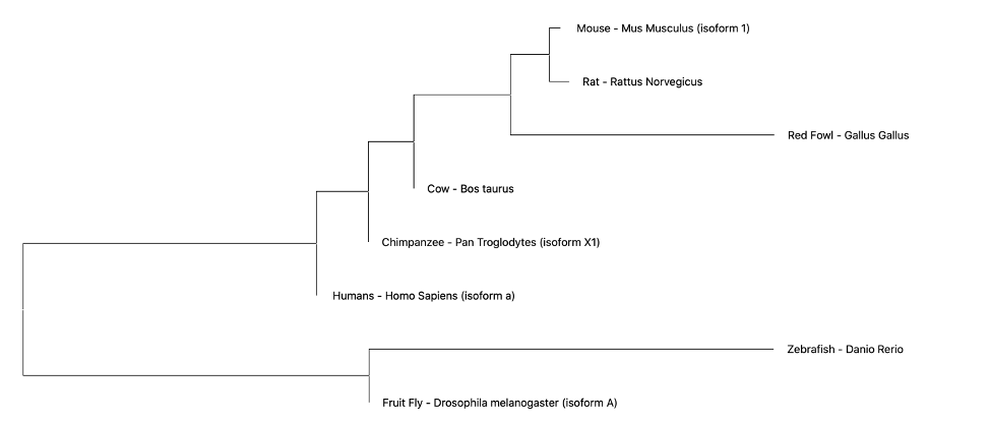
Discussion
Model organisms determined from phylogeny and homology may be useful for EGRF research; namely EGFR-Induced Lung tumor mice can be studied to determine the invasive phenotype [4]. Taking into consideration the cost and requirements to grow these organisms should also contribute to this decision. Furthermore, the fact that primates are the most homologous could be important to EGFR's role in lung physiology in the context of EGFR.
References:
1.) Khan Academy, Building a phylogenetic trees. Retrieved from: https://www.khanacademy.org/science/biology/her/tree-of-life/a/building-an-evolutionary-tree
2.) Saitou N1, Nei M, The neighbor-joining method: a new method for reconstructing phylogenetic trees (1987). Retrieved from: https://www.ncbi.nlm.nih.gov/pubmed/3447015
3.) E. A. Thomson, The method of minimum evolution (1973). Retrieved from: https://onlinelibrary.wiley.com/doi/abs/10.1111/j.1469-1809.1973.tb00595.x
4.) Politi, Fan, et al., Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down-regulation of the receptors (2006). Retrieved from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1475762/
2.) Saitou N1, Nei M, The neighbor-joining method: a new method for reconstructing phylogenetic trees (1987). Retrieved from: https://www.ncbi.nlm.nih.gov/pubmed/3447015
3.) E. A. Thomson, The method of minimum evolution (1973). Retrieved from: https://onlinelibrary.wiley.com/doi/abs/10.1111/j.1469-1809.1973.tb00595.x
4.) Politi, Fan, et al., Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down-regulation of the receptors (2006). Retrieved from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1475762/
